


Introduction
Renal insufficiency and failure are characterized by multiple and complex disturbances in mineral and bone metabolism. In the past decade, the term chronic kidney diseaseŌĆōmineral and bone disorder (CKD-MBD) was developed to describe a ŌĆ£syndromeŌĆØ which is not exclusively restricted to bone metabolism. In this context, Block et al [1] showed the relevance of increased serum phosphate levels Ōēź5.52┬Āmg/dL (1.78┬Āmmol/L). In 1998, in a large retrospective observational study of hemodialysis patients, such a level of hyperphosphatemia was shown to be associated with a significant increase in death from cardiovascular disease [1]. Later, at the turn of the millennium, an intensive discussion started after the introduction of oral phosphate binders which were not based on calcium regarding the adverse potential of calcium loading of the body as a synergistic trigger of morbidity from cardiovascular disease [2], [3], [4]. Since then, both the stigmata of CKD-MBD, hyperphosphatemia and a positive calcium balance, have been accepted as key inductors of the initiation and progression of cardiovascular calcification in CKD.
Historical background
Mortality increases in the early stages of CKD at a creatinine clearance Ōēż60┬ĀmL/min [5], [6]. As we have learnt that vessel calcification is not just a simple passive process of calciumŌĆōphosphate precipitation, but is a consequence of modified gene expression with the active induction of phenotype transformation of smooth muscle cells into osteoblasts within the vessel wall [7], [8], attention on this process has increased and concentrated on the basic control and regulatory mechanisms involved [9], [10]. The aim of this research is to reduce the potentially lethal sequelae of disturbed homeostasis in mineral metabolism in CKD [11]. Multiple and recent epidemiological studies have documented associations between ionic and humoral abnormalities on the one side, and morbidity and mortality on the other [12].
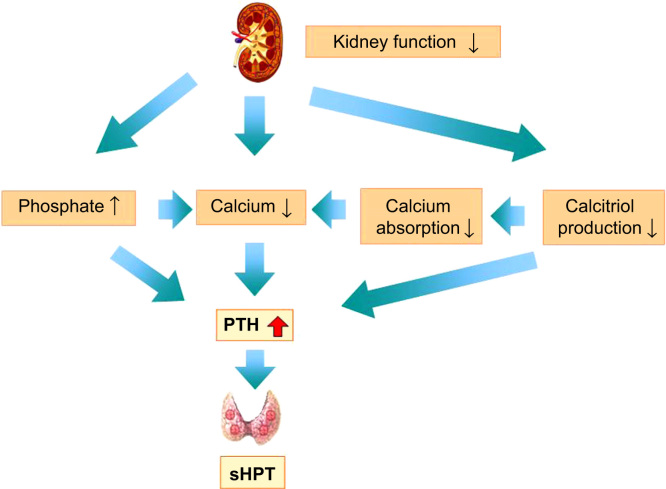
It was initially believed that progressive fibrosis of the kidneys with a loss of normal parenchymal tissue was the functional cause of decreasing excretory function (recognizable by reduced urine production and solute clearance). It was also believed that this was the underlying cause of the progressive decrease in incretory capacity which results in reduced levels of endogenous active vitamin D and compensatory increases in parathyroid hormone (PTH) levels to ward off imminent hypocalcemia (Fig. 1).
In addition, following the discovery of highly potent, protective hormonal mechanisms which induce an increase in phosphaturia (the so-called phosphatonins) and which thus attenuate the development of hyperphosphatemia, the original concept of an exclusive vitamin DŌĆōhypocalcemia perception in the development of CKD-MBD has been modified over the last decade to include a primarily phosphate regulated and regulating paradigm (Fig. 2) [13].
Recently, the COSMOS (Current Management Of Secondary Hyperparathyroidism ŌĆō a Multicenter Observational Study) study results showed a 22% reduction in all-cause mortality and a 29% reduction in cardiovascular mortality in patients treated with phosphate binders. The open cohort, observational study consisted of 6,797 patients followed prospectively for 3 years in 227 dialysis centers from 20 European countries. Remarkably, the reduction in mortality was also shown in patients treated with calcium-based phosphate binders, whereas more marked reductions were noted in patients treated with combinations of phosphate binders [14].
Phosphate control mechanisms
The discovery of the phosphatonins, including fibroblast growth factor 23 (FGF-23) [15], [16] and Klotho [17] allowed new insights into the pathogenesis of CKD-MBD. FGF-23 consists of 251 amino acids with a molecular weight of 26,000┬ĀDa and is produced primarily in osteocytes [18]. As yet, the exact mechanisms which result in its secretion have not been completely elucidated; however, it is generally accepted that increased phosphate loading or hyperphosphatemia directly or indirectly stimulates FGF-23. In addition, calcitriol stimulates the secretion of FGF-23, and FGF-23 is bound into feedback loops which also suppress the secretion of PTH and calcitriol [19], [20], [21].
FGF-23 can be detected via its intact and C-terminal sequences, although, at present, certain differential diagnosis cannot be deduced from the two assay targets. Remarkably, FGF-23 values can increase by a factor of more than 1,000-fold in end-stage renal disease, which can, at least in part, be interpreted as a weakening of the feedback loops and, in the case of C-terminal assays, as cumulation in CKD. The production and sensitivity of the receptor-coactivator Klotho is downregulated in CKD as it is also under the direct negative influence of FGF-23. Furthermore, Klotho expression is partially dependent on calcitriol, which is progressively reduced in CKD [22], [23].
In the presence of Klotho, FGF-23 binds to FGF receptors, utilizing a dimeric receptor complex to induce specific signal transduction. FGF receptors are detectable in most organs; however, the coexpression of Klotho is specific to the kidneys and parathyroid glands [24].
FGF-23 suppresses the expression of the sodiumŌĆōphosphate (NaPi) cotransporters NaPi-2a and NaPi-2c in the proximal renal tubules and augments phosphate excretion [25], [26]. The original name ŌĆ£KlothoŌĆØ (derived from Greek mythology) illustrates the high expectations regarding new insights, as decreasing Klotho levels in CKD could possibly explain the premature aging of multiple organ systems. In fact, the discoverer of the Klotho gene describes phosphate as the signaling molecule of aging [27].
Remarkably, Isakova et al [28] were able to show that an increase in FGF-23 is detectable early in the development of CKD, although serum phosphate levels were either low or normal in these stages of CKD. At this point it remains speculative and simultaneously still plausible that the increase in FGF-23 could be interpreted as a protective mechanism against phosphate overload in a scenario of already decreasing renal clearance, although the exact trigger mechanism of this regulatory process remains to be identified.
Calcimimetics reduce not only PTH, but also FGF-23 [29]. Chonchol et al [30] showed, in a double-blind randomized, placebo-controlled study of 404 patients in CKD stages 3 and 4, that treatment with cinacalcet suppressed PTH levels in these patients. However, this positive effect was accompanied by an increase in phosphate levels, which was probably caused by a reduction not only in PTH, but also in FGF-23.
Isolated reduction of FGF-23 results in an increase in serum phosphate. This was shown by Shalhoub et al [31] in a study of rats with CKD which were treated with antibodies specific for FGF-23. Partial normalization of bone parameters, i.e. PTH and calcitriol, and osseus structure were observed; however, a marked increase in serum phosphate, vessel calcification, and mortality were also seen. Thus FGF-23 and PTH, in their roles as phosphatonins, are indispensable in producing an adequate reduction in renal phosphate reabsorption from initially 80ŌĆō90% to approximately 15% in advanced renal failure [32] and therefore in a reduction in deleterious cardiovascular sequelae of phosphate loading. These results confirm the hypothesis that evolving hyperparathyroidism in the early stages of CKD may have to be regarded primarily as a beneficial, adaptive mechanism supporting the homeostasis of normal serum phosphate levels and loads.
Data presented so far suggest that FGF-23 is the parameter which increases first in CKD-MBD [33]. However, FGF-23 can apparently induce left ventricular hypertrophy and therefore potentially increase cardiovascular risk [34], [35]. Activation of renin-angiotensin-aldosterone system (RAAS) or the induction of inflammation have also been suggested as relevant pathomechanisms [36]. Therefore FGF-23 should not be viewed as an isolated laboratory parameter. Depending on the circumstances, FGF-23 is on one hand physiologically protective, but, on the other hand, pathophysiologically detrimental under extreme conditions [37]. It currently remains obscure whether the interpretation and differentiation of adaption or maladaption is acceptable on the basis of the absolute size of FGF-23 levels or if we can assume a relatively sharp distinction between maintained kidney function and terminal end-stage disease with the obligatory requirement of dialysis.
At present, there is no simple picture of FGF-23 regarding monocausal associations and therapeutic aspects; FGF-23 correlates with the progression of renal failure [38] and numerous studies have identified an independent association between FGF-23 and all-cause or cardiovascular prognosis [39], [40], [41], although the quantitative prognostic power of FGF-23 in manifest end-stage renal failure is inhomogeneous at the moment [42], [43], [44].
Interpretations of early phosphate reduction studies in CKD
Surprisingly, and contrary to the expectation of a reduction in vessel calcification in CKD stages 3bŌĆō4 by the attenuation of the phosphate load as a result of treatment with oral phosphate binders, Block et al [45] showed the progression of vasculature calcification in a placebo-controlled, direct comparison of phosphate binders, which was especially associated with calcium-based phosphate binders. Although phosphate excretion in urine was reduced by 22%, levels of PTH and FGF-23 remained substantially unchanged. Although the number of patients was relatively small, with approximately 40 patients in each of the four groups in the study, these results highlight our incomplete understanding of this pathophysiology [46]. This specific cohort did not have overt hyperphosphatemia, but had serum phosphate levels just in the high normal range, showing a reduction from 1.36┬Āmmol/L (4.2┬Āmg/dL) in both the active and placebo groups to 1.26┬Āmmol/L (3.9┬Āmg/dL) with active treatment and 1.32┬Āmmol/L (4.1┬Āmg/dL) for the group treated with a placebo. Furthermore, FGF-23 was not highly stimulated overall (mean 223┬ĀRU/mL). However, iPTH was moderately increased [mean approximately 80.4┬Āpg/mL (8.52┬Āpmol/L)].
Differing reductions in FGF-23 while treated with various phosphate binders highlight the complexity of these counter-independent relations, with no significant reduction with calcium-based binders compared with larger reductions in FGF-23 with calcium-free binders in patients on hemodialysis [47], [48]. Furthermore, a study by Hill et al [49] of eight patients in CKD stages 3b and 4 with average phosphate levels of 3.8┬Āmg/dL (1.23┬Āmmol/L) showed a neutral calcium and phosphate balance on an average diet containing approximately 1,000┬Āmg calcium and 1,500┬Āmg phosphate per day. Utilizing calcium carbonate as the phosphate binder, this metabolic study confirmed a small but significant reduction in phosphaturia, while iPTH and FGF-23 remained unchanged. The administration of calcium carbonate lead to a positive calcium balance, while, and again unexpectedly, active intervention did not affect the total phosphate balance.
At present we have insufficient data to recommend a target range for FGF-23. In addition, there is no indication to determine FGF-23 in daily clinical routine. However, it appears plausible to evaluate an FGF-23-guided treatment approach in the future and to favor treatment options which modulate the biological mechanisms physiologically. At the moment, attention should focus on the selection of phosphate binders, the differentiated utilization of calcimimetics and diligent administration of vitamin D or a selective vitamin D analog, accounting for bone status dynamics. The future challenge is the recognition of optimal parameter combinations to further adapt treatment options.
Conclusions and future perspectives
With respect to the question ŌĆ£Treatment of phosphate retention ŌĆö the earlier the better?ŌĆØ the results of the study of Hill et al [49] possibly suggest that there is no early retention of phosphate. However, a repeat study with a phosphate binder not based on calcium may allow further insights into the mechanisms involved because the calcium content of the phosphate binder may have confounded the results. According to the study of Block et al [45], treatment with phosphate binders alone in the early stages of CKD appears to have no persuasive cardiovascular advantages, but patients at risk should possibly be identified in the future by using FGF-23 ranges, which have not yet been defined.
The validity of these assumptions are supported by the recently published DOPPS (The Dialysis Outcomes and Practice Patterns Study) results [50], showing a 25% lower mortality [hazard ratio (HR) 0.75; 95% confidence interval (CI) 0.68ŌĆō0.83] in patients prescribed phosphate binders when adjusted for serum phosphate level and other covariates. Further adjustment for nutritional indicators attenuated this association (HR 0.88; 95% CI 0.80ŌĆō0.97). However, this inverse association was only observed for patients with serum phosphate levels Ōēź3.5┬Āmg/dL (Ōēź1.13┬Āmmol/L). In the instrumental-variable analysis, the facility percentage of phosphate binder prescription adjusted for case mix was associated positively with a better nutritional status and inversely with mortality (HR for 10% more phosphate binders 0.93; 95% CI 0.89ŌĆō0.96). Further adjustment for nutritional indicators reduced this association to an HR of 0.95 (95% CI 0.92ŌĆō0.99), thus showing once again the complexity of interactions between phosphate loading and corporeal handling on one hand, and the often overlooked aspect of nutritional status on the other; this also questions the validity of the COSMOS findings.
We also need to test the parallel (synergistic) administration of NaPi-2b receptor inhibitors [51]. FGF-23 inhibits NaPi-2b receptor activity and thus reduces intestinal phosphate absorption [52], [53]. This option appears attractive as an in vitro analysis of rats has shown that >90% of active phosphate absorption is facilitated by the NaPi-2b transporter [54], which is intestinally upregulated while receiving standard treatment with phosphate binders or strict dietary phosphate restriction [55]. Therefore the regulation of the NaPi-2b transporter plays a significant part in phosphate homeostasis [53].
Excessive calcium supplementation appears to be deleterious with respect to cardiovascular integrity. Indeed, a recently published high quality meta-analysis of 11 randomized studies consisting of 4,622 patients showed a 22% reduction in total mortality for calcium-free versus calcium-based phosphate binders (risk ratio 0.78; 95% CI 0.61ŌĆō0.98) [56]. Furthermore, direct and specific antagonism of FGF-23 effects in early CKD should be avoided.
In our opinion, the indication for treatment should, for the time being, not be based on a supposed and potentially false assumption of inadequate phosphate retention, but on the presence of visible hyperphosphatemia until the basic interdependent mechanisms in CKD have been elucidated in more detail.
Conflicts of interest
Patrick Biggar has received honoraria for speaking and advisory tasks from AbbVie, Amgen, Sanofi/Genzyme, Ineos Health Care, Fresenius Medical Care, Medice, Hexal, and Takeda. Markus Ketteler has received honoraria for speaking and advisory tasks from AbbVie, Amgen, Fresenius Medical Care, Medice, Mitsubishi, Sanofi, Shire, and Vifor, and has received research funding from AbbVie and Amgen. Samuel K.S. Fung has declared no conflict of interest.

 PDF Links
PDF Links PubReader
PubReader Full text via DOI
Full text via DOI Download Citation
Download Citation Print
Print






")









